|
根室側の研究成果 日時:平成18年1月26日(木)〜1月27日(金)
I. ハナサキガニ資源の遺伝的多様性と集団構造に関する研究
東北大学大学院集団遺伝情報システム学分野:研究者代表者:谷口順彦
研究者:池田 実・アンナ バリノワ・吉村友香理
1. 目的
昨年度までに、ハナサキガニ資源の遺伝的多様性を把握するための遺伝マーカーとして高変異性のマイクロサテライトDNAの増幅プライマーセットを7種類開発した。これらのプライマーセットを用いることにより、根室半島沿岸域、南千島(クリルスキー)、およびサハリンにおけるハナサキ集団の遺伝的多様性について検討した(昨年度報告書参照)。この結果の妥当性について確認するためには、マイクロサテライトDNAのような核DNAだけではなく、核外のミトコンドリア(mt)DNAも対象として検討を行う必要がある。昨年度はmtDNAマーカーとしてCytochrome oxidaseサブユニットI(CO I)の部分配列を用いたが、変異量が低く、集団間の遺伝的分化について詳しい解析を行うことができなかった。今年度は、集団解析に有用な多くの突然変異がCO Iよりも多く蓄積されていると考えられる調節領域(A+T rich領域)を同定し、遺伝マーカーとしての評価と確立を試みた。その上で、この領域の配列情報を用いて上記3海域の集団遺伝学的解析を行い、マイクロサテライトDNAから得られた結果との比較検討を行うことを目的とした。また、新たにカムチャッカの集団も加えて、同様の分析を行い、3海域の集団との遺伝的関係についても検討を行った。
2. 成果
1)ハナサキガニ調節領域の同定と集団遺伝マーカーとしての評価
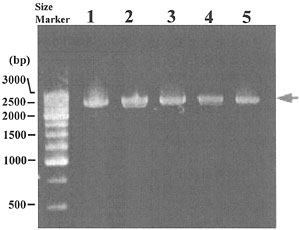
非コード領域である調節領域をPCRにより効率良く増幅するためには、調節領域ならびに近傍の遺伝子配置ならびにこれらの塩基配列を把握しておくことが必要である。しかし、ハナサキガニを含むタラバガニ科については、調節領域の塩基配列はおろか、近傍の遺伝子配置すら明らかにされていない。一方、異尾類のうちタラバガニ科と最も近縁なのはホンヤドカリ科であることが形態および一部の遺伝子の再配列データから提出されており、その一種Pagurus longicarpusではmtDNAの全塩基配列が明らかにされている。そこで、この種の配列情報を積極的に活用することとした。まず、調節領域がP. longicarpus同様にCyt b遺伝子とND I遺伝子に挟まれていると仮定してプライマーをデザインし、PCRを行ったところ約2.5kbpの増幅断片を得ることに成功した(図1)。このPCR断片をテンプレートとして、プライマーウオーキングにより断片の全塩基配列を決定し、ホモロジーサーチおよびtRNAの二次構造予測等により遺伝子配置の推定を行った。
図1. 調節領域が含まれていると考えられるPCR断片
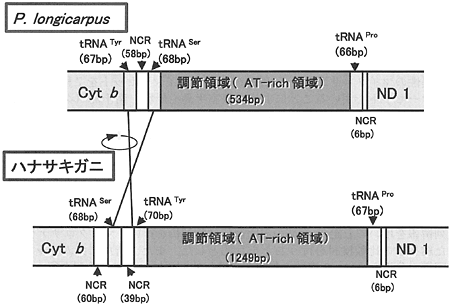
その結果、図2に示すように、基本的な配置はP. longicarpusと同様であったが、tRNATyrとtRNASerの位置が入れ替わっており、さらにtRNATyrの配列が逆転していた。調節領域はtRNATyrにtRNAProに挟まれた1249bpと推定されたが、予測されたORF(オープンリーディングフレーム)が既知のmtDNAの遺伝子配列と全く一致しないこと、AとTの割合が他の節足動物での調節領域の値とほぼ同程度であったことから、調節領域とみなして差し支えないと考えられた。
| 図2. |
ホンヤドカリの1種(P. longicarpus)とハナサキガニにおける調節領域近傍の遺伝子配置の比較 |
どの程度の変異が調節領域に蓄積されているかを検討するため、珸瑤瑁の標本集団34個体について調節領域前半部の359塩基の配列を調べた。その結果、表1に示すように30サイトで変異が検出された。このうち塩基置換が起きていたサイトは25箇所、欠失または挿入が起きていたサイトは6箇所で、30個のハプロタイプが分類された。調節領域の変異量とCO I領域の変異量を比較した結果、調べた個体数は異なるものの、ハプロタイプ数で約3倍、ハプロタイプ多様度(h)で約1.5倍、塩基多様度(π)で約5倍となり、CO Iよりも著しく高い変異量を示した(表2)。この結果から、調節領域の塩基配列はハナサキガニ集団の遺伝的多様性を詳細に検討する上で極めて有効なマーカーになりうることが示された。
表1. 珸瑤瑁の34個体で検出されたハプロタイプと変異サイト
表2. 珸瑤瑁集団におけるCO Iと調節領域の変異量の比較
|
対象とした
サイト数 |
調べた
個体数 |
変異
サイト数 |
ハプロ
タイプ数 |
ハプロタイプ
多様度(h) |
塩基多様度
(π) |
| CO I |
332 |
50 |
9 |
11 |
0.684 |
0.003 |
| 調節領域 |
358 |
34 |
30 |
30 |
0.993 |
0.017 |
|
2)カムチャッカの集団を含めた遺伝的多様性の再評価
(1)mtDNA分析
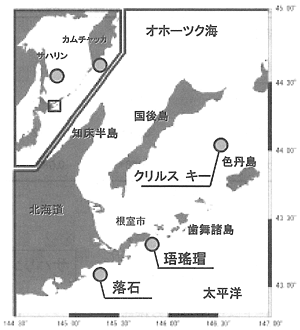
図3に調べたハナサキガニ標本集団の採集位置を示す。珸瑤瑁、落石、南千島(クリルスキー)、サハリンは昨年度に検討したサンプルと同一である。これらはそれぞれ32〜38個体、カムチャッカの標本集団は42個体を調べた。各標本集団の調節領域における変異性を比較した結果、根室半島沿岸や南千島ではハプロタイプ多様度が1に近いのに対して、サハリンやカムチャッカではそれぞれ0.845と0.904で、これらの海域での集団の遺伝的変異性が低いことが示された。この傾向は、出現ハプロタイプの分散を示す有効ハプロタイプ数で顕著に現れており、カムチャッカやサハリンでは特定のハプロタイプの出現頻度が他の海域に比べて高いことが示された。
図3. ハナサキガニ標本の採集海域
表3. ハナサキガニ5集団におけるmtDNA調節領域の変異性の比較
| 集団 |
個体数 |
ハプロタイプ数 |
有効ハプロ
タイプ数 |
ハプロタイプ
多様度(h) |
塩基多様度
(π) |
| 落石 |
38 |
26 |
17.6 |
0.969 |
0.017 |
| 珸瑤瑁 |
34 |
30 |
27.5 |
0.993 |
0.017 |
| クリルスキー |
34 |
29 |
22.4 |
0.986 |
0.016 |
| サハリン |
32 |
13 |
5.5 |
0.845 |
0.017 |
| カムチャッカ |
42 |
21 |
8.5 |
0.904 |
0.017 |
|
標本集団間の遺伝的分化の有無について検討するため、調べた標本集団全体での分化程度をAMOVA分析により検討したところ、標本集団間の差異によって説明される遺伝的変異の割合は2.94%と0よりも有意に大きい値を示し(P=0.0011)、明らかに遺伝的分化が生じていることが示された。次にどの海域間で分化が生じているのかを検討するため、標本集団間でのFST(遺伝子分化指数)を計算した結果を表4に示す。根室海域および南千島とサハリンとの間で有意なFST値(0.085〜0.100、P<0.005)が得られ、全体の遺伝的分化のうち大部分がこれらの海域間の差異によってもたらされているものと考えられた。また、根室海域と南千島の間ではいずれの組み合わせにおいても実質的にFST=0とみなされる負の値を示した。このことは、昨年度のマイクロサテライトDNA分析によって得られた結果を支持するものであり、三角海域でのハナサキガニの遺伝的組成は均質とみなすのが妥当と考えられる。サハリンの次に低い変異量を示したカムチャッカと他の海域間では有意な値は得られなかったが、FST値自体は0.012〜0.017と三角海域間での値に比べて高いものであった。
表4. ハナサキガニ5集団間のFST値
|
落石 |
珸瑤瑁 |
クリルスキー |
カムチャッカ |
サハリン |
| 落石 |
- |
|
|
|
|
| 珸瑤瑁 |
-0.015 |
- |
|
|
|
| クリルスキー |
-0.009 |
-0.009 |
- |
|
|
| カムチャッカ |
0.015 |
0.012 |
0.016 |
- |
|
| サハリン |
0.096* |
0.085* |
0.100* |
0.017 |
- |
|
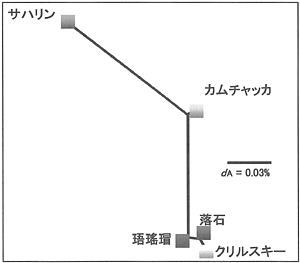
遺伝的類縁関係について検討するため、標本集団間の純塩基置換率を求め、近隣結合法によって類縁図を作成した。その結果、根室海域と南千島は同一のクラスターを形成し、サハリンとは大きく離れた関係となった。また、カムチャッカはこれらの間に位置した(図4)。
図4. mtDNA分析によるハナサキガニ5集団の遺伝的類縁関係
|