|
INTRODUCTION
This lecture will focus on advances in three major neurologic diseases that reach their maximum in persons older than 65 years: stroke, Parkinsonism and Alzheimer's disease. I discuss them not only because they affect such a large number of patients but also because each represents a distinct kind of brain illness, thereby illustrating the varying challenges that face present-day neuroscientific research into etiology and therapy. Stroke, although mostly resulting from a primary disease of the arteries, underscores the elderly brain's singular vulnerability to oxygen lack or external injury. Recent epidemiologic discoveries about stroke emphasize the great beneficial effect that preventive measures can confer on the aging brain. Parkinsonism serves as an example of an unexplained degenerative disease of the nervous system which sometimes can result from external forces; it exemplifies how the environment and the natural predisposition of the individual may interact to create brain disease. Thirdly, I'll discuss Alzheimer's disease, also a degenerative brain disorder which is rapidly becoming the single most common and socioeconomically devastating cause of chronic medical disability in the industrial world.
1. STROKE
Let me turn my discussion first to stroke. Stroke stands second only to Alzheimer's disease in frequency in both our countries. Furthermore, it provides an example of how much epidemiologic research on preventive measures can reduce the incidence of a geriatric disease by delaying its onset.
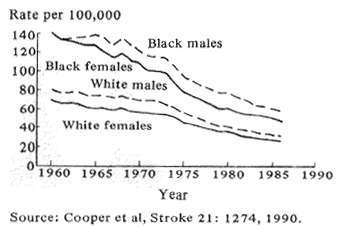
Fig. 1 Trends in age adjusted stroke mortality U.S. 1960-86
Stroke incidence in the U.S. started to fall during the 1950's and continues to remain substantially less frequent in relation to age than was the case 40 years ago (Fig. 1; Cooper et al, 1990). This favorable change can be attributed to three very different scientific approaches. First, epidemiologic analyses of vascular problems, particularly the control of hypertension, along with changing dietary habits and acting on the results of controlled treatment trials have had a large impact in reducing stroke incidence and, therefore, mortality. Second, newly discovered drugs which dissolve acute clots, called thrombolytic agents, show promise in preliminary trials of being able to reverse or reduce the damage of acute stroke; and third, recent advances in brain pharmacology and biochemistry suggest that drug therapy given after a stroke beings may be able to reduce the danger to the brain of the associated oxygen lack. Let me expand briefly on these points.
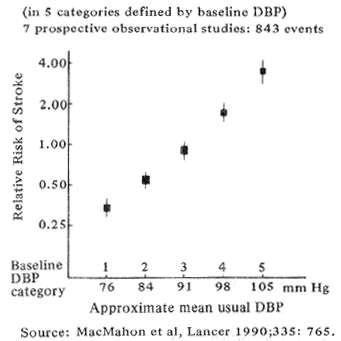
Fig. 2 Stroke and usual diastolic blood pressure (DBP) in a large epidemiologic meta analysis
Epidemiologic studies beginning as long as forty years ago first suggested that chronic hyper-tension bears a strong causative relationship to stroke. The association has recently been re-affirmed by MacMahon's (1990) meta-analysis of 420,000 persons shown in Fig. 2. The data demonstrate a direct relation between stroke and diastolic blood pressure, equalling an 8-fold increase in stroke risk between pressure of 80 and l05. As part of the same study, Collins et al, (1990), showed that no matter how severe the hypertension, therapeutically lowering the mean blood pressure by as little as 6 mm Hg reduced the odds of future strokes by approximately 42%.
Epidemiologic research similarly has demonstrated that pharmacologic prophylaxis reduces stroke risk. A summary analysis of large clinical trials conducted on 29,000 patients (Antiplatelet Collab., 1988) in North America and Europe found that the platelet inhibitor, aspirin, can effectively reduce the risk of developing secondary, but not primary stroke. This means that the drug reduces stroke risk only after a previous heart or brain attack. The protective effect amounted to a significant 27%. Other studies comparing aspirin to the newly introduced, more expensive, antiplatelet agent, ticlopidine, indicate that it may give a slightly greater degree of secondary stroke prevention, amounting to as much as 30% (Gent, 1989; Haas, 1989). Somewhat tempering the small advantage, is the finding that ticlopidine may cause a serious neutropenia in one percent of its users.
Epidemiologic research also has demonstrated forcefully that cigarette smoking predisposes to stroke and that cessation can reduce stroke risk almost as much as does treating hypertension. Two large American analyses, one performed by Wolf (1988) as part of a long term study of the Framingham, MA. population the other by Colditz (1988) on a large cohort of nurses evaluated for a total of over 900,000 patient-years, found that moderately heavy cigarette smoking at least doubles stroke risk for men and increases it by 350% for women. At least within the framework of five year follow-up studies, abstinence eliminated the increased risk within two years of withholding cigarettes.
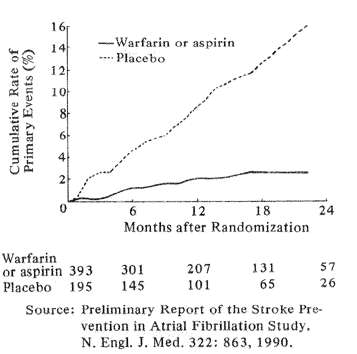
Fig. 3 Rates of stroke or systemic embolism (primary events) in patients given Warfarin or Aspirin (solid, lower curve) or placebo (dotted upper curve)
Recent large, controlled clinical trials have found anticoagulants to have proved value in preventing stroke in several high-risk situations. Although past analyses established a strong role for anticoagulants in chronic atrial fibrillation due to rheumatic heart disease, only recently have large cooperative studies from Denmark and the USA shown that warfarin or, in some instances, warfarin or ASA significantly reduces stroke risk from chronic, nonrheumatic atrial fibrillation. The Danish study (Peterson et al, 1990) showed that warfarin but not aspirin significantly reduced stroke risk from chronic atrial fibrillation. A North American study (Stroke Prevention in Atrial Fibrillation Study Group Investigators 1990) found that in patients younger than age 75 years, either warfarin or aspirin reduced stroke risk from atrial fibrillation by over 80%. The respective value of ASA and warfarin has not yet been determined in this study, but here again aspirin had no effect on persons older than age 75. Fig. 3 graphically describes the number of stroke or stroke-like episodes that occurred in the placebo group compared to the ASA-warfarin fibrillators over the first 24 months of study.

Table 1 TPA in acute stroke
Let me now turn to recent clinical research on thrombolytic agents. These are aimed at dissolving intra-arterial emboli or thrombi so as to prevent brain damage. A first, NIH sponsored, open trial of tissue plasminogen activator (t-PA) has given encouraging results among 90 patients treated within three hours of the onset of sustained stroke symptoms. None of the patients were hypertensive, all had normal clotting mechanisms and none showed evidence of cerebral hemorrhage by pretreatment CT-scans. Forty percent of the 90 patients treated within three hours showed substantial improvement both clinically and by CT measured infarct size (Table l). Furthermore, among 74 patients treating within 90 minutes of onset, 20 showed almost immediate, dramatic improvement. Four patients suffered associated cerebral hemorrhages; one was fatal, and three others died as a direct result of stroke. Both clinical outcomes and death rates in this preliminary trial were significantly better than predicted from historical controls in the Cornell data bank. Since stroke outcome can vary widely among differently selected population groups, controlled scientific trials by NIH are now underway to test these first impressions.
Much research has gone into analysis of the cell biology of stroke during the past few years with results that have stimulated several new approaches to treatment (Plum and Levy, 1991).
Following cerebral arterial thrombosis or cardiac arrest, oxygen lack shuts down the mitochondrial energy factory in brain cells in a very short time. Nevertheless, a variety of studies indicate that if the circulation can be restored within less than an hour, much if not most of the otherwise expected brain damage can be prevented. Even later restoration of flow up to as much as two to three hours after onset may reduce the size of the stroke. Except when thrombolytics can be quickly injected, establishing arterial reflow is neither technically easy nor widely practical because of the narrow time window during which it must be given.
Faced with the above problem, investigators have carried out a number basic experimental studies in tissue culture and various animal species to examine four major areas of brain cellular vulnerability during ischemia (Choi, 1990). These include 1) the development and testing of calcium channel blockers, aimed at improving tissue blood flow and preventing calcium from entering and poisoning nerve cells; 2) The development of neuronal receptor blockers directed at preventing excitatory neurotransmitters from beating vulnerable neurons to death; 3) Providing buffering against potentially lethal lactic acidosis in nerve cells during and after ischemia and 4) scavenging or deionizing free oxygen atoms which otherwise could initiate potentially fatal tissue peroxidation. Each of these approaches has yielded promising results but none, so far have been proved to have a major effect on clinical stroke.
2. DEGENERATIVE BRAIN DISEASES
I now turn to recent neuroscience research on the late life degenerative brain diseases, choosing the major examples of Parkinson's and Alzheimer's diseases. At the outset, let me explain to those in the audience who are unfamiliar with neurology that when the brain and especially the human brain shows degenerative changes, they affect very selective parts of the structure. Man's brain is biologically a far more complicated organ than it might first appear. The brain is far from being a single homogeneous piece of tissue such as, for example, the heart, the lung or the liver. Rather, the compact brain, an instrument which ultimately controls all aspects of the body's mind and executive functions, represents nature's astonishingly efficient way of packaging into a small protected space, no larger than a melon, a large number of anatomically and chemically different subcomponents. Indeed, the total of those specialized brain subunits is greater than the number of separate countries placed on the face of Asia. Imagine! One brain system sees, another smells, still another hears, while still more function to regulate balance, to maintain temperature, to think, to speak and to learn. Furthermore, large integrating circuits must pull all these functions together to act as a unit. Each of the brain's functional subcompo nents, or systems as they often are called, develops according to specific rules set out by its particular guiding genes. Each decays gradually according to its intrinsic genetically preordained program and each often has a special chemical vulnerability or resistance to a selective group of potentially lethal internal or external toxins or other threats. Most degenerative diseases of the central nervous system, reflecting a vulnerability dictated by their selective genetic development, attack only a limited number of the brain's or spinal cord's systems. As a result, although the natural deterioration of advancing age serves as a common denominator to several geriatric neurologic probr lems, the beginning stages of most degenerative diseases produce unique clinical features and singular pathologies. Presently, so little is known about the general principles that initiate these features, that scientists who wish to solve the mysteries of primary brain diseases have little choice but to attack them individually, hoping that answers found to one may more broadly answer some of the questions that arise in others as well. Illustrating this principle, common neuropathologic abnormalities can occur between Parkinson's and Alzheimer's diseases but double dividends of this kind are unusual. Molecular neuroscience still stands a long way from understanding how and why certain subsystems of the brain are more susceptible than others to the ravages of time.
3. PARKINSON'S DISEASE
Parkinson's Disease, at least in its early stages, reflects the premature, progressive functional insufficiency and death of a tiny population of melanin-containing nerve cells lying in the deeply located substantia nigra of the upper brain stem. The affected neurons manufacture the catecholamine neurotransmitter dopamine, sending the transmitter along its output fibers, called the nigrostriatal tract to reach the striatum. The latter is part of the basal ganglia, a set of large subcortical brain nuclei which coordinate and modulate normal voluntary movement. In the striatum, dopamine serves a damping function, acting as a modulatory, inhibitory transmitter.
Neither the etiology, the mechanism of cell death nor why Parkinson's disease inexorably progresses are well understood.An important consideration, however, is that by its late stages the abnormalities in many cases of Parkinson's disease extend and overlap those of Alzheimer's disease.

Table 2 Parkinson's disease-predisposing or causative factors
Like Alzheimer's, Parkinson's disease shows a modest familial incidence although genetic factors seem not to be paramount since identical twins have no higher a susceptibility than dizygotic ones (Table 2). Unlike Alzheimer's, however, several known exogenous conditions can precipitate Parkinsonism. Some readers may recall the outbreak of Parkinson's disease that gradually emerged for nearly 40 years following the influenzal epidemic of the 1920's, suggesting that viral agents could in some way cause or predispose to the illness. Epidemiologic studies have not identified any similar incidents since that time so that the particular agent must have been biologically unique. Moderately severe anoxia especially when prolonged for several hours can induce signs and symptoms of Parkinsonism but such patients usually have gross infarcts of the striatum allowing little precision in identifying cellular mechanisms of the disease.
Several years ago, very small amounts of an illegally produced street drug of the tetrohydropyridine family called MPTP were found to duplicate almost exactly in humans the anatomic and pharmacologic abnormalities of severe Parkinson's disease. The discovery created great interest as a possible clue to the wider causes of Parkinson's disease. Research showed that the brain converts MPTP via the enzyme monoamine oxidase B into a phenylpyridine called MPP. MPP, in turn, enters the mitochondria of nigral dopamine neurons in which it inhibits respiration and allows the entry of lethal concentrations of calcium. Scientists have proposed that other environmental toxins may similarly enter brain to act on substantia nigra and other vulnerable areas. But the problem of explaining the progression remains. Some evidence, gained from clinical trials suggests that selegeline, which inhibits amine oxidase B may slow the natural breakdown of dopamine, thereby prolonging its action, and also may protect nigral cells against environmental toxicity. Otherwise, extensive searches for MPTP-like chemicals in the environment have been fruitless although knowledge of the poison has made it possible to develop excellent animal models of the disease on which future therapy can be tested.
Several scientists have tried to replace in the brain of MPTP-damaged monkeys or patients with natural Parkinsonism, new cells to replace the dead or dying nigral neurons. Initial efforts consisted of attempts to homo-transplant pieces of the patient's or animal's own adrenal gland tissue into the diseased basal ganglia. These experiments have produced variable and, in patients, sometimes fatal outcomes. As a result, adrenal transplants have greatly declined in clinical use. Recently. Lindvall, Bjorkland and their Swedish colleagues (1990) have conducted meticulously designed experiments, transplanting bits of human fetal substantia nigra into the dopamine deficient putamen of two patients with Parkinsonism. Early studies suggest that the transplants survive, make connections and proceed to synthesize the missing enzyme. At least, one of the patients, has enjoyed impressive clinical and pharmacologic improvement, with positron emission tomography showing increased dopamine receptor activity in the basal ganglia. Obviously, this experiment requires verification in other cases. Also, the ethical concepts of transplanting human fetal material into the brain undoubtedly will receive much discussion. Whatever the outcome, this experiment provides a strong stimulus to finding ways to facilitate cellular regeneration in the brain and develop practical measures for enhancing nerve cell transplants into that organ. Our Cornell laboratories (Goldman, 1990) are working vigorously on the problem of inducing vertebrate brain neurons to survive and multiply in tissue culture. Already, we have determined, that with the help of certain neurotrophic factors to which I return later, we can stimulate nerve cells from vertebrate brains to subdivide in tissue culture.
One dreams of a future day when it will be possible to obtain on a large scale successive division and multiplication of specific kinds of nerve cells in tissue culture and can then use those cells to replace dying neuronal populations in the large numbers of patients who need them.
4. ALZHEIMER'S DISEASE
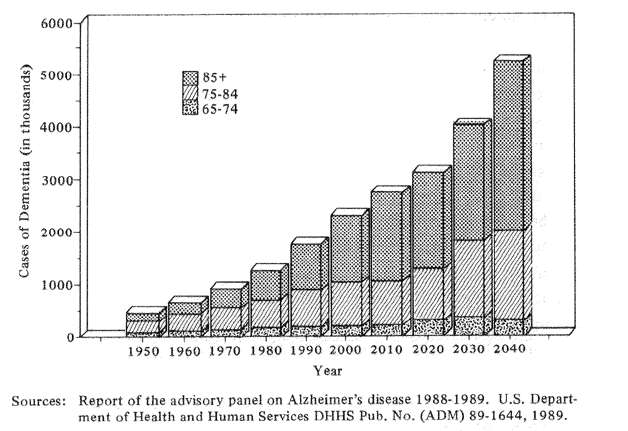
Fig. 4 Estimated numbers of cases of severe dementia by age in the U.S. population, 1950-2040
I turn now to Alzheimer's disease, a new plague which threatens to overwhelm the health care and social service systems of the rapidly aging industrial world. The problem is huge. In the United States of 1980 approximately 1.3 million persons were calculated as being incapacitated by Alzheimer's Disease and related dementias. By 1990, the number of Alzheimer cases had increased to approximately 1.8 million or 38% in a single decade (Fig. 4). If we extrapolate these numbers by presently known health and survival statistics, Americans can expect an increase in incapacitating dementia by the year 2040 of approximately 400% over the 1980 figures. The social and economic implications are staggering not only for North Americans, but for the Japanese and North European communities as well.
Unless we find effective prevention, 50 years from now the costs of Aizheimer's disease in suffering and financial costs threaten to exceed those of all other health care needs combined.
The name, Alzheimer's disease, describes a specific neuroanatomic selective degeneration of certain brain cells accompanied by fairly typical mental changes in a still relatively healthy body. Alzheimer's may or may not be a disease, as such. What is certain is that the condition is linked inseparably to aging, since similar but quantitatively less severe behavioral and morphological changes can be found in most elderly persons as well as in lower mammals including baboons and even rats. This cross-species resemblance has great importance since it allows basic research on the disease to extend beyond the test tube into animal models.
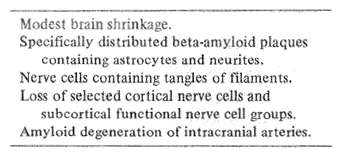
Table 3 Anatomy of Alzheimer's disease
Anatomy is the road map of neuroscience because it gives the first hint of a disorder's predilection to involve a specific brain system or systems. Alzheimer's disease is accompanied by four major anatomic abnormalities (Table 3). These include: 1) Plaques containing a special kind of protein, called beta amyloid. These especially involve the association areas of the frontal and parietal lobes, as well as the deeper hippocampus and amygdala; 2) Visible abnormalities in nerve cell fibrils, producing a tangled appearance; 3) Neuronal dropout in critical cortical areas and subcortical anatomic nuclei, and 4) Beta amyloid degeneration affecting intracranial blood vessels, the full significance of which has so far been little explored.
Brain scientists are vigorously persuing each of the above major anatomic abnormalities as possibly holding a key to the pathogenesis of the Alzheimer's problem. I'll return to these issues in a moment. Meanwhile, let me spend a moment about possible etiology.

Table 4 Proposed etiologies of Alzheimer's disease
Presently, only a minimum of clues point to the possible cause of Alzheimer's disease (Table 4). Environmental factors appear relatively unimportant inasmuch as the disorder has a roughly similar age-related incidence worldwide. Genetic factors establish in advance a natural program of cell growth and death which undoubtedly plays a major role. This principle of programmed cell death is best illustrated by changes during early normal brain development, but it occurs at all ages. The mammalian brain in young fetuses sprouts out far more nerve cells than it eventually will use. Inherent, genetically governed programs weed out the excess, progressively honing and tuning neural networks by the time of birth to meet their future, naturally selected demands. Eventually, however, the "weeding out" process, so necessary for normal development becomes humanity's biological enemy. As time plods on, the years thin out phylogenetically newer parts of the nervous system but the visceral housekeeping parts survive. Although the rate at which they die is disputed, all human brains are genetically programmed to lose nerve cells progressively after about the 25th year of life. The brain cannot naturally replace these nerve cell losses anymore than a bald head can naturally grow new hair. But even if the process of aging plays a part in Alzheimer's, other mechanisms must set the timing for when the disease appears.

Table 5 Genetic factors in Alzheimer's disease (AD)
Strong evidence suggests a genetic influence in Alzheimer's (Table 5). Population studies indicate that fully half of the first degree relatives of patients with AD will develop the disease if they live to the age of approximately 85 years. The number is at least 15% higher than among similarly aged controls without such relatives but even the latter figures implies a universal, inherent predisposition. Despite this impressive evidence for genetic factors, intuitively known by most physicians who deal with the aged and their families, identification of a specific Alzheimer's gene or genes has proved elusive. Initially, geneticists postulated that the offending gene might lie on chromosome 21 because 100% of persons with Down's Syndrome, a condition associated with a triplication of the 2lst chromosome, developed the neuroanatomic changes of AD by age 30. Subsequently, identification of an apparently abnormal chromosomal marker on CR 21 in certain families with well documented autosomal dominant, presenile Alzheimer's appeared to support the hypothesis. The chromosomal error, however, has not held true for other elderly patients and families with the disease. What does seem to be important is that the gene responsible for synthesizing a protein called the amyloid beta protein precursor lies on chromosome 21. A fragment of this precursor makes up the core of the Alzheimer plaques as well as the amyloid that resides in intracranial blood vessels, both these abnormalities being major markers for both aging and the disease.

Table 6 Alzheimer amyloid beta protein (ABP)
Major scientific effort has focused on brain amyloid in the pathogenesis of AD (Table 6). The amyloid precursor protein (APP) that appears so abundantly in Alzheimer's has been strongly conserved in evolution and is found within several different body tissues among a large variety of animal species, (Selkoe, 1990). The finding implies that the precursor protein possesses strong functional properties as yet unclarified. The amyloid pre cursor protein has an unusual configuration which straddles cell membranes leaving a smaller end within the cell while the larger one dangles outside. Additionally, the larger end sometimes contains an insert which turns out to be a known protease inhibitor. Certain molecular sequences of the extracellular part of the amyloid precursor molecule suggest that it could contribute to wound healing processes or even serve as a growth factor. Whichever the case, amyloid beta protein precursor is a normal functioning protein in humans but appears in excess in patients with Down's Syndrome and the brains of patients with autosomal dominant Alzheimer's disease.
In both normal and abnormal aging, an excess of beta amyloid in the brain precedes the development of the full scale Alzheimer plaque. Furthermore, both plaques and tangles appear in the brains of 50-75% of persons over the age of 70 years who show no evidence of dementia; the big difference is that the number of these changes is less in normals than in typical AD. Presently, I find it difficult to assess whether the degraded amyloid protein in Alzheimer plaques plays a primary rather than a secondary, mop ping up role in AD. Evidence from Down's Syndrome, familial AD with a 21 chromosomal defect and late life normals suggests that the quantity of brain amyloid relates directly to the protein's gene action on chromosome 21 but only secondarily to AD. Whether the abnormal ABP in the brain emanates from blood vessels or neurons themselves and whether or not the protein is in any way neurotoxic remains to be determined.
Neurofibrillary tangles, demonstrated as twisted paired helical filaments within the brain's large pyramidal neurons, comprise a second, prominent pathological marker of AD. Long at issue has been whether the tangles reflect a primary abnormality of the disease or, alternatively, consist of a degenerating group of intracytoplasmic proteins, representing secondary responses in already dying cells. The fact that similarly tangled cells occur in other neurologic degenerations including Pick's disease, progressive supra-nuclear palsy and, some forms of amyotrophic lateral sclerosis suggests that their presence is a secondary response to an earlier, more specific abnormality. Recent evidence indicates that at least part of their tangled paired helical filaments consists of an abnormal variation of normal brain protein called tau rather than a newly generated, disease-specific chemical.
Neuronal degeneration and selective death of several specific brain neuronal systems provide an important part of the defined neuropathology of AD. In AD, scattered losses of large pyramidal cells containing neurofibrillary tangles or granulovacuolar degeneration appear in several cortical and subcortical areas, most prominently including the amygdala, hypothalamus and hippocampus as well as the frontal and parietal association cortex. Prominent early areas of cell loss affect the cholinergic basal forebrain nuclei and, to a lesser extent, the noradrenergic locus ceruleus and serotonergic dorsal and superior raphe nuclei of the brain stem.
Although their causative role remains controversial, anatomic studies emphasize that these rostrally aimed subcortical autonomic nuclei frequently degenerate in Alzheimer's disease. The first systematic neurochemical studies of Alzheimer brains by Bowen et al (1976) and Davies and Maloney (1976) demonstrated a marked selective decline in acetylcholine receptors in the human cerebral and limbic cortex. Shortly afterwards Whitehouse, et al (1982), described in postmortem Alzheimer brains extensive cell loss in the area of the cholinergic basal forebrain and septal nuclei. These deep nuclei project widely and diffusely to the cerebral cortex. It was already known in humans that pharmacologic cholinergic blockade with atropine-like drugs sometimes caused memory loss and confusion, especially in elderly persons. A variety of studies in animals similarly demonstrated impaired memory in association with degeneration or destruction of basal forebrain lesions (Bartus et al, 1982).
Even when the mechanistic causes of a disease remain incompletely understood, doctors nevertheless try urgently to find measures that may bring symptomatic relief. That principle for example, led to repeated unsuccessful efforts to treat Parkinsonism until Cotzias finally reaches success using gradually increasing, huge doses of D-L DOPA. Unfortunately analogous efforts to treat human AD with cholinergic drugs or their congeners have brought out, at best, only minimal clinical improvement. Animal experiments, however, suggest that other strategies might produce more favorable results.
Nilsson and his co-workers (1990) provide such an example. They first determined that when they destroyed the cholinergic basal forebrains and brainstem serotonergic nuclei in young rats, the animals developed impaired memory and poor spatial searching strategies with difficulties similar to those found in aged rodents. When the investigators grafted appropriate cellular replacements for the missing neurotransmitters into the hippocampus, the animals returned to a normal performance whereas controls never recovered spontaneously. For a variety of evident reasons, these experiments are interesting, but the transplanting of human or even non-immunogenic animal cells into the brain can hardly provide a practical, numerical solution to solving the increasing epidemic of degenerative neurologic diseases. A direction that some neuroscientists think might help lies in exploiting the potential for the naturally sustaining influence of neurally generated growth or trophic factors to slow down or halt the neuronal decay that underlies these devastating conditions.
5. NEUROTROPHIC FACTORS AND THE AGING BRAIN
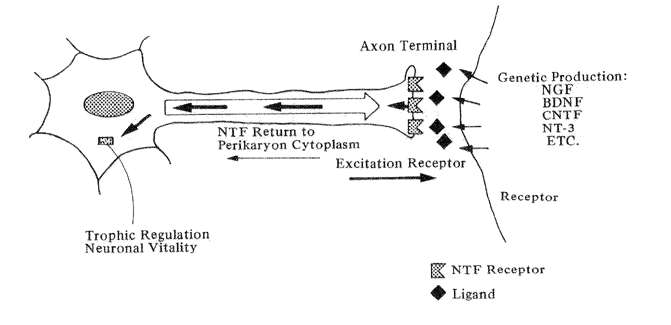
Fig. 5 Diagrammatic example of the actions of neurotrophic factors (Orthodromic excitation from the axon terminal stimulates the release of neurotrophic factor(s) from the receptor which travel back up the axon to affect the nerve cell body.)
The guiding hypothesis of research in neurotrophic factors in that aging-induced decay in nerve cells may not necessarily reflect a primary deterioration in their intrinsic mechanism but may instead be caused by a loss of reverse stimulation from their target cells (Snider and Johnson, 1989). The concept is illustrated in Fig. 5: a neuron's discharging axon stimulates the receptors of its targeted nerve cell. The latter cell, in turn, release a neurotrophic agent back across the synapse which is picked up by specific receptors on the parent cell. The molecule then travels back up the axon to up regulate lysosomal genes in the cytoplasm which maintain the vitality of the first order neuron.

Table 7 Neurotrophic factors
Growth or trophic factors have several known, important functions (Table 7). They promote neuron survival during development. They maintain the specific function of mature nerve cells. They enhance the regeneration of cut nerve fibers. They are highly specific for particular populations of central and peripheral nervous system neurons and they are normally present in neural tissues in only tiny amounts. But, they have great power to maintain the survival of nerve cell systems.
Factors of this kind have been demonstrated indirectly since 1950 when Victor Hamburg and Rita Levi Montalcini did their earliest experiments on nerve growth factor. The factor itself was directly isolated by 1960, then specifically characterized in the 70's and 80's in steps that led to a Nobel Prize for Levi Montalcini and Sidney Cohen. During the past few years, scientists have identified several new molecules that possess major trophic qualities.
Thus far, five major neurotrophic growth factors have been identified by their tissue source. They include Nerve Growth Factor (NGF), Brain De rived Neurotrophic Factor (BDNF), Ciliary Neurotrophic Factor (CNTF), Neurotrophin-3 (NT 3) and Fibroblast Growth Factor (FGF).
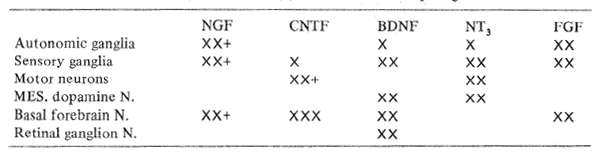
Table 8 Known in vitro (X) or in vivo (+) effects of neurotrophic growth factors
The particular targets of the major neurotrophic factors are compared in Table 8. Nerve growth factor, NGF, has a variety of effects in tissue culture, acting most strongly on peripheral autonomic ganglia. NGF in experimental rats has been shown by Fischer et al (1986), to provide protection against degenerating basal forebrain cells and memory impairment in elderly animals. In addition, Apfel et al (1990), have demonstrated that NGF protects mice against the severe peripheral nerve damage otherwise caused by anticancer chemotherapy.
Brain Derived Neurotrophic Factor (BDNF) enhances the survival and protection of developing mesencephalic dopaminergic and basal forebrain cholinergic neurons as well as spinal motor neurons in tissue culture. If one draws an analogy from experience with NGF, these qualities suggest it could have protective effects in the adult against senile memory loss. Parkinsonism and, possibly, amyotrophic lateral sclerosis.
Ciliary Neurotrophic Factor (CNTF) enhances the development in culture of a variety of neuron types. Recently CNTF has been shown by Sendtner et al, to protect against retrograde facial nucleus degeneration in animals with proximal facial nerve sections, again encouraging future experimental trials in motor neuron degenerations.
Neurotrophin 3 (NT 3) is the most newly discovered and characterized polypeptide growth factor with amino acid sequences similar to both NGF and BDNF. Thus far, the agent has been found to promote the survival and out-growth of both central and peripheral embryonic neurons in tissue culture. Its further properties remain to be determined by experiment.
The findings in tissue culture and experimental animals suggest that brain-derived neurotrophic factors may eventually offer a measure of treatment to patients with Alzheimer's disease, Parkinsonism, Amyotrophic lateral sclerosis and acute toxic neuropathy.
Before clinical tests of neurotrophic factors can be conducted, however, much must be done.. Since all these factors represent naturally occurring, peptides that are highly conserved across animal species, they are not likely to stimulate serious adverse immune responses. Nevertheless, other kinds of adverse reactions remain highly possible: no one knows what complications may ensue in either the nervous system or systemic tissues when long-nascent neural enzymes suddenly are turned on late in life. Furthermore, all of these agents have a macromolecular size that seriously impedes their passage across the blood brain barrier to reach their desired targets. All of these issues will require time and meticulous attention to evaluate. But, given the helplessness and hopelessness that presently surrounds the victims and families of Alzheimer's disease, Parkinsonism, amyotrophic lateral sclerosis and therapy-needing cancer, it is highly likely that these precautions to therapeutic trials will be rapidly faced and dealt with. We and others already are working with such issues but as matters look presently, it seems likely that carefully restricted first human trials of NGF may soon become possible.
REFERENCES
l) Antiplatelet Trialist Collaboration: Secondary prevention of vascular disease by prolonged antiplatelet treatment. Brit Med J, 296: 320-331 (1988).
2) Apfel SC, Lipton RB. Arrezo JC, Kessler JA: Neuronotrophic factor rescue of toxic neuropathy. Ann Neurol, 28:220 (1990).
3) Bartus RT, Dean RL, Beer B, Lippa AS: The cholinergic hypothesis of geriatric memory dysfunction. Science, 217:408-417 (1982).
4) Bowen DM, Smith CB, White P, Davison AN: Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain, 99:459-496 (1976).
5) Choi DW: Cerebral hypoxia; some new approaches and unanswered questions. J Neurosci., lO:2493-2501 (1990).
6) Colditz GA, Bonita R, Stamfer MJ, et al: Cigarette smoking and risk of stroke in middle aged women. N Engl J Med., 318:937-941 (1988).
7) Collins R, Peto R, MacMahon S, et al: Blood pressure, stroke and coronary artery disease. Part 2. Short term reductions in blood pressure: overview of randomized trials in their epidemiological context, Lancet., 335:827-838 (1990).
8) Cooper R, Sempos C, Shih-Chao H, et al: Slowdown in the decline of stroke mortality in the United States, 1978-1986. Stroke., 21:1274-1279 (1990).
9) Davies P, Maloney AJF: Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet, 2:1403 (1976).
lO) Fischer W, Wictorin K, Bjorklund A, et al: Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature, 329:65-68 (1987).
11) Gent M. Blakely JA, Easton JD, et al: The Canadian-American ticlodipine study (CATS) in thromboembolic stroke. Lancet, l:1215-1220 (1989).
12) Goldman S: Neuronal development and migration in explant cultures of the adult canary forebrain. J Neurosci, lO:2931-2939 (1990).
13) Haas WK. Easton JD, Adams HP, et al: A randomized trial comparing ticlodipine hydrochloride with aspirin for the prevention of stroke in high risk patients. N Engl J Med, 321:501-507 (1989).
14) Lindvall O, Brundin P, Widner H, et al: Crafts of fetal dopamine neurons survive and improve motor function in Parkinson's disease. Science, 247:574-579 (1990).
15) MacMahon S, Peto R, Cutler, et al: Blood pressure, stroke and coronary artery disease. Part 1. Prolonged differences in blood pressure and prospective observational studies corrected for the regression dilution bias. Lancet, 335:765-774 (1990).
16) Nillson OG, Brundin P, Bjorkland A: Amelioration of spatial memory impairment by intra hippocampal grafts of mixed septal and raphe tissue in rats with combined cholinergic and serotonergic denervation of the forebrain. Brain Research, 516:193-206 (1990).
l7) Peterson P. Boysen G, Godfredson J, et al: Placebo controlled randomized trial of warfarin and aspirin for prevention of thromboembolic complications in chronic atrial fibrillation. Lancet, l:175-179 (1989).
18) Plum F, Levy DE: Preventive and acute therapy of established benefit in stroke. In: J Krieglstein, H Oberpickler (eds), Pharmacology of Cerebral Ischemia; Wissenschaftliche Verlagsgesllschaft mbH, Stuttgart (1991).
19) Selkoe DJ: Deciphering Alzheimer's disease: the amyloid precursor protein yields new clues. Science, 248:l058-l06O (1990). 20) Snider WD, Johnson DM: Neurotrophic molecules. Ann. Neurol., 26:489-506 (1989).
21) Stroke Prevention in Atrial Fibrillation Study Group Investigators: Preliminary report of the stroke prevention in atrial fibrillation study. New Engl J Med, 322:12 (1990).
22) Whitehouse PJ. Price DL, Stuble RG, et al: Alzheimer's and senile dementia: loss of neurons in the basal forebrain. Science, 215:1237-1239 (1982).
23) Wolf PA, D'Agostino RB, Kannel WB, et al: Cigarette smoking as a risk factor for stroke: The Frarningham study. JAMA, 259:1025-1029 (1988).
|
